![]() Link
to Zeiss M63 Photomicroscope Tutorial
Link
to Zeiss M63 Photomicroscope Tutorial![]()
Introduction 
Although the light microscope is the most commonly used biological instrument, it is often used improperly. This may not matter so much with very thin commercial slides but proper alignment of the illumination system is essential for viewing thick sections and whole mounts. It is also crucial for photomicroscopy. You will be using microscopes throughout this class and for years to come. If you learn the simple lessons we will teach you today you will do much better in your work and see the exciting world of microscopy in a new light! (Sorry about the pun). The procedure we follow was developed by the German scientist, August Kohler (1866-1948), and it bears his name. Recently his ideas were used to make an advanced Electron Microscope by Zeiss. Thus, this procedure which was introduced in 1893 has been of lasting and value.
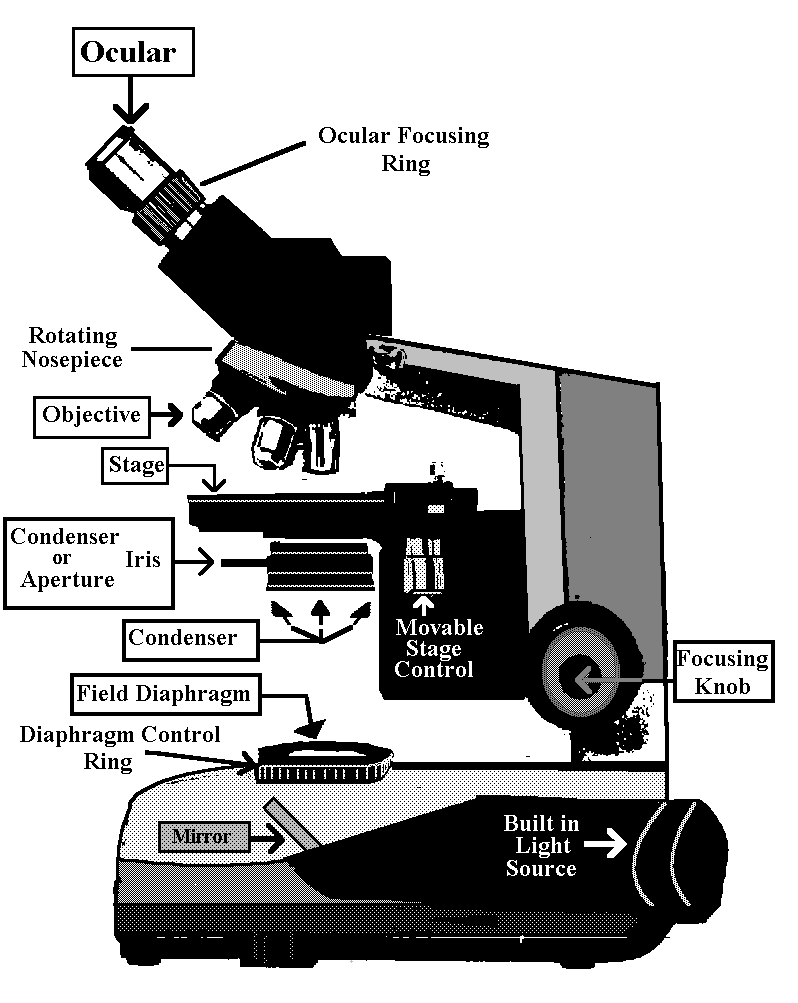
The Compound Microscope
Because the lens systems in a microscope are composed of many lenses it is called compound. The typical illumination of specimens in which light passes through the specimen and travels to your eye is called Bright Field microscopy. Light has the following path.
Eye or Camera
Ocular ->
Body or Tube ->
Specimen ->
Objective ->
Condenser (Iris) ->
Condenser Lenses ->
Field (Iris) Diaphragm ->
Mirror ->
Light Source ->
Locate these parts of your microscope by referring to Illustration above.
We will be using Leitz microscopes in this class, however, the instructions for correct alignment of the condenser will be applicable to other microscopes with adjustable condensers.
Light is provided by a built in bulb which is reflected through the field iris diaphragm, the condenser, the condenser iris diaphragm, the specimen, the objective, the tube and the ocular.
There are various control knobs on the microscope which affect the light path. In addition, there are knobs for coarse and fine focus, as well as knobs to move the stage.
Locate the coarse and fine focusing knobs on each side of your scope. Each knob does coarse and fine focusing. There is no separate knob for fine focusing. You will see how this works later. The rotation of this knob focuses the objective onto the specimen.
Mechanical Stage
The knobs which control the mechanical stage are on the right side of the microscope as it faces you.
Condenser
The condenser aligns and focuses 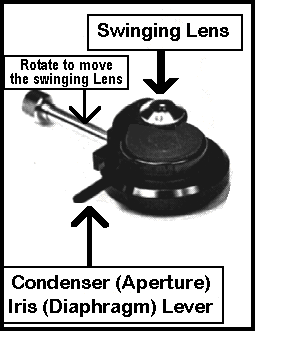 light on the specimen. It has a long vertical
knob [pointing down] on its left side as you face the scope. Rotation of the small knob at
its tip, raises and lowers the condenser to achieve focus.
light on the specimen. It has a long vertical
knob [pointing down] on its left side as you face the scope. Rotation of the small knob at
its tip, raises and lowers the condenser to achieve focus.
Swinging Lens
Directly above this at a
right angle to the condenser adjusting knob you will find a rod which controls a lens
which can be swung in or out of place. This swinging lens is left out for low-power
illumination [i.e. 4X], and swung into the light path for objectives of 10X or greater
magnification. Failure to use this lens properly is the most
common mistake that most people make. If you fail to use this properly you will not
be able to see much, especially when we use thick sections or whole mounts.
sections or whole mounts.
Centering Screws
In addition, there are two small knobs on the front of the condenser, set at 45o which are used to center it.
Aperture Iris Diaphragm
Finally, there is a lever which
controls the aperture iris. This improves contrast (difference between light & dark)
especially at intermediate and high magnifications.
Do NOT use this to increase or decrease brightness!!!
Locate all of the controls for the condenser.
REMEMBER THIS !!!!!!!!!!!!!!!!!!!!!!!!!! |
|
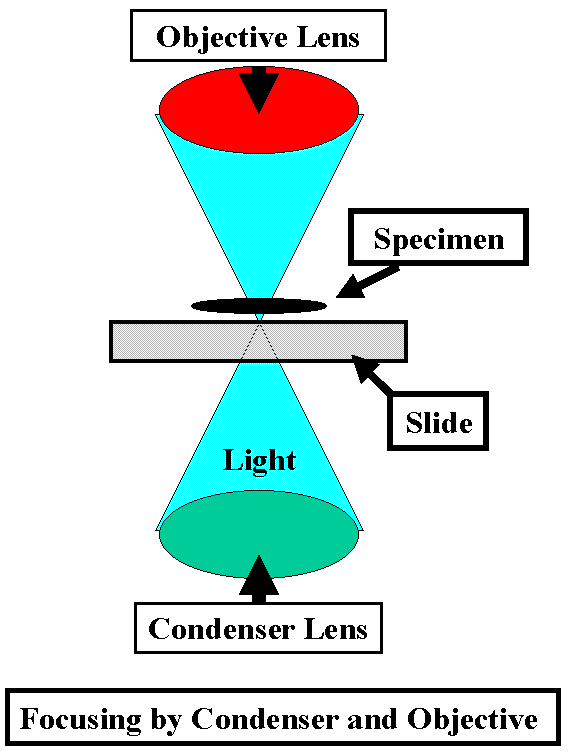
The Objective
focuses on the Specimen
The Condenser focuses light onto the |
|
Field
Diaphragm
The light source is housed in the base of the microscope. It passes through the field iris diaphragm. The size of the field diaphragm is controlled by rotating a knurled ring which is concentric with it. The field diaphragm controls the area of illumination.
Locate the field diaphragm and its knurled ring.
Objectives
The magnification of an image is
primarily controlled by the objectives which are housed in a rotating nose piece. To
change objectives you rotate the nosepiece, starting with the 4X objective. Do not start
viewing by swinging in the 20 - 100 X objectives. These may be damaged if they hit the
specimen.
The magnification is indicated by a number on each objective. Furthermore there is a
progression in size such that the longest objectives have greatest magnification. The
distance between the objectives and the cover slip (working distance) decreases
dramatically as the magnification of the objective increases.
The 100 X objective is an oil
immersion lens. Note the black line near the tip of the objective. This is used to
identify an oil immersion lens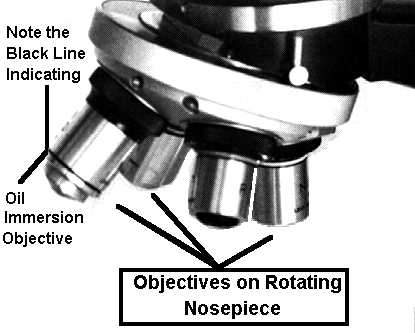 .
.
Place a small drop of oil on the objective lens.
A small drop of oil must also be placed on the cover slip.
The lens should be carefully lowered
into the oil prior to focusing.
Observe this with your naked eyes focusing on the objective and the specimen. Do NOT look through the oculars.
Oil improves the optics because it unites the glass cover slip and the objective. It
replaces air with oil. The oil has the same refractive index as glass. Thus less light
scattering & refraction occurs.
Be sure that the specimen was in focus at 40X before switching to 100X.
Avoid focusing down on the specimen with an oil immersion lens. Change the focus so that
the objective is traveling away from the slide. If the image does not come into focus,
reverse the direction until it does. When in doubt, STOP!!! & ask for HELP!!!
The lens might be dirty or there may be some other problem.
The oil also protects the objective lens from scratching.
Notice that we have 4, 10, 20, 40 & 100 X objectives. Always start with the 4 X objective to prevent damage to the other objectives which may collide with the specimen.
Once one of the lenses is focused an
a specimen, the others should also be in focus when they are swung into place. This
property is referred to by the term parafocal. However, in actual practice some adjustment
is required when you switch from one objective to another. This usually presents little
difficulty. However, you must be especially careful when switching from 10X to 40X and
from 40X to 100X.
We will often be using fresh sections and whole mounts in the class. These can be thick
and irregular. Consequently, greater care must be taken when changing objectives. When in
doubt, play it safe  and ask for help until you get
acquainted with the material you are studying.
and ask for help until you get
acquainted with the material you are studying.
Oculars
The oculars should be adjusted to
suit both of your eyes. Note that there is a scale on the tube holding both objectives. We
will label microscopes so that each student can work with the same instrument throughout
the course.
Grasp the adjustable knurled ring below each ocular with your thumb and forefinger and
gently rotate it so that each is set at 64 which is its midpoint.
Before you make any adjustments, place a slide on the stage and focus on part of the
specimen.
You are now ready to achieve Kohler illumination. Oh happy day!
Kohler Illumination
The best resolution occurs when all elements of the microscope are in perfect alignment and the iris diaphragms are properly adjusted to the best aperture. On simple microscopes you may not be able to alter the alignment of the different parts, but on these Leitz microscopes it is possible to align and focus the condenser to achieve "Kohler Illumination".
Because we will be using a lot of thick hand-sections in this class, it is vital that you learn how to achieve Kohler illumination. Otherwise, you will not be able to analyze your specimens.
1] Place a commercially prepared slide on the stage.
2] Make sure the swinging lens is in
the light path (facing up) and focus on the specimen using the 10X objective.
3] Use only one eye [right eye with right ocular or left eye with left ocular] and focus the specimen with the coarse/fine focusing knob.
4] Use the knurled ring below the other ocular to focus it while looking through it with your other eye. You may not need to change the focus. However, experiment by rotating the knurled focusing ring to see its effect. My German friends have told me that the correct way to focus the second ocular is to make it more negative so it is out of focus, then rotate it in a positive direction until it is focused.
5] Having the oculars focused will
improve image quality and will decrease eye strain. Once this is done it need not be changed during a given session. However, it is a good habit
to do this at the beginning of each lab. It is best done at 10X because there is less
chance for errors at this magnification compared to 4 X.
6] Make sure the aperture iris is completely open [rotated all the way counter-clockwise].
7] Reduce the field of illumination
by rotating the knurled ring on the field diaphragm completely
clockwise. Be gentle with the field diaphragm. It should close without any effort.
8] You should see a small circle of light. If you are lucky, it will be in the center of the field. However, it will most likely be off-center and out of focus. Let us know if you can't find it!!
9] Use the vertical condenser adjusting knob to make the circle as small as possible by gently rotating it. This moves the condenser up and down. Do this carefully so that the circle of light is not pushed laterally. As you focus the field diaphragm you will notice that its halo turns from blue to red and red to blue. The best focus occurs when you adjust the condenser so that the halo is just between red and blue. This is a little hard to do so don't be too worried if you have some red or blue in the halo.
10] Expand the field diaphragm by
rotating its knurled ring counter-clockwise, until the light touches one edge of the
field. If the light is perfectly centered it should touch the entire circumference of the
field. This is unlikely.
11] Center the circle of light by using the two small adjustable knobs on the front of the condenser. When you are satisfied, expand the field so that the light fills it completely. However, do not fully open the field diaphragm. Open it just enough to extend beyond the field of view.
12] Repeat this with the 20 or 40 X objective. For critical work this should be done for each objective. This is especially important for taking photographs and for examining minute, translucent specimens like fungi and algae. For our labs, it will be good to do this for the 10X objective at the start of each session. You need not do this for 4X and 40X. However, if you are having some problems resolving details, check to be sure that you have the condenser aligned and focused.
It may be difficult to do this with the 100 X objective. However, if you achieve proper alignment with the 40 X objective, the 100 X will be similar.
13] When working at 20 - 100 X it is
important to adjust the condenser aperture iris. This is especially important for
translucent structures. Closing this iris increases contrast. Thus something fuzzy becomes
smooth and something faint becomes dark. It is usually possible to close the iris and
judge its effects subjectively. However,  there
is a "tried & true" procedure which you should know.
there
is a "tried & true" procedure which you should know.
14] Remove one of the oculars and
look directly down the tube at the light field. Close the iris so that it occludes 1/4 -
1/3 of the area. This should give the best contrast. Examine a specimen before and after
adjusting the aperture iris. This should be done for each objective for critical viewing.
In practice, you can experiment with this while viewing a specimen and adjust it without
removing the ocular. Closing the aperture iris also increases depth of focus up to a
point. Thus, more areas of a three dimensional specimen will be in focus If it is closed
to much, a flat indistinct image results.
The example shows part of a diatom frustule. There is little detail when the iris is wide
open (top). When it is fully closed (middle) the contrast is increased but there are
aberrations which make the small holes appear larger than they are in actuality. The
outline of the small holes is also indistinct. When the iris is closed 25 - 30 % there is
improved contrast and less aberration.
15] Experiment with the aperture iris while viewing a prepared slide. Once you have achieved what you think gives the best image quality, remove one of the oculars and see how much of the field is occluded.
As part of the first lab, we will be using different stains to study their effects an fresh specimens. Experiment with the aperture iris as you study these. Fresh sections are usually too thick for detailed examination at high magnification, but the aperture iris can be used to great effect with this type of material.
While these procedures may seem tedious, they will become routine as you progress in the course.
Hand Sections
The ability to make free hand sections will allow you to quickly analyze plant organs without resorting to laborious procedures. A tremendous amount of information can be derived from hand sections. These do not need to be extremely thin to be of use. In addition, hand sections of a structure do not need to be complete or uniformly thin to be useful. Your initial attempts at hand sectioning will probably be frustrating, however, you will quickly become proficient. Hand sections also provide 3-D information which is not available with most commercial slides.
Instructions (Right-Handed)
1] Place a Band-Aid on the thumb of your left hand. Have the cotton portion on the bottom of your thumb. The thumb is a backstop for this operation.
2] Place another on the end of your index finger. The index finger will control the height go the specimen, and thus its thickness.
3] Grasp the plant structure between your thumb and forefinger so that the top of the specimen extends above the level of your forefinger.
4] Take a single-edge razor blade in your right hand. Be sure that it is wet.
5. Rest the blade on your forefinger
and use a 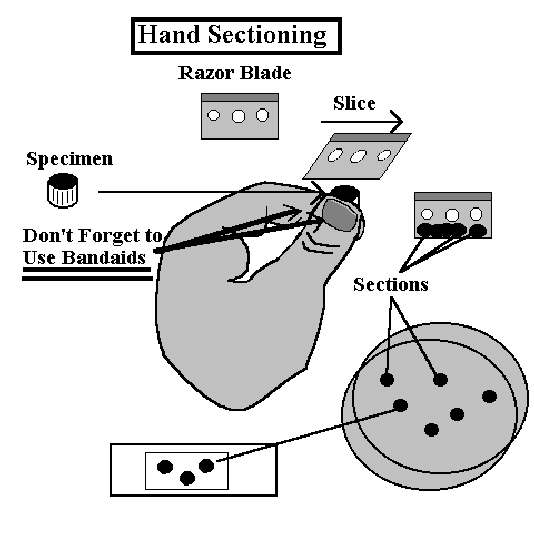 slicing motion to cut off the top of the specimen.
slicing motion to cut off the top of the specimen.
6. Try to avoid cutting your thumb with the blade!!!
7] Raise the specimen slightly by manipulating it with your fingers and repeat the slicing motion.
8] Thin sections can often be obtained by pressing the blade down on your forefinger and then slicing through the specimen several times.
9] After several sections have accumulated on the blade, wash them off in a Petri dish of water.
10] Keep on slicing until you have some thin sections. These will appear translucent when seen against the dark background of your lab bench. In most cases, the sections will have thin and thick regions. As long as part of the section is thin, you may be able to use it, and thick sections are frequently OK.
11] Sections can be removed with forceps and placed in a drop of water or stain on a microscope slide.
12] It is a good idea to view unstained sections prior to staining. Proper use of the aperture iris is important for this.
Adding
Coverslips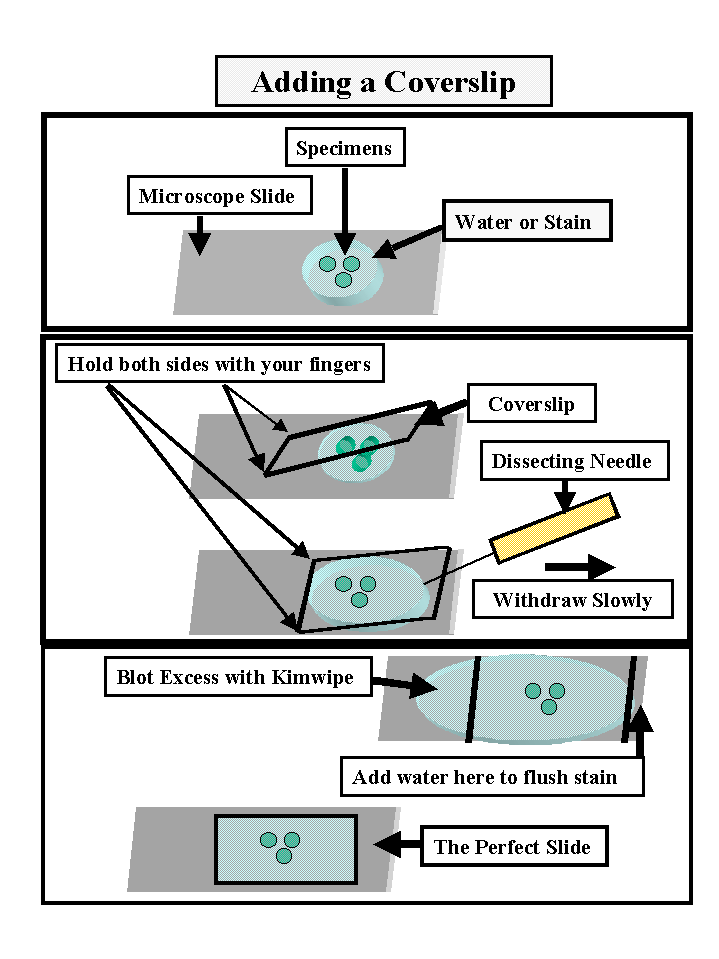
It is essential that the sections be completely immersed in water so that air is excluded.
Air bubbles or spaces will interfere greatly with your observations. To avoid these when adding a coverslip do the following.
a] Place your sections in 2-3 drops of water or stain in the center of the slide.
b] Use a fine forceps to pick up a large cover slip (20 x 40 or 20 x 50)
c] Place one end of the coverslip on the slide (near boundary with frosting) without touching the solution containing the specimens.
d] Steady this end with the fingers of your left hand.
e] Slowly lower the forceps until it touches the slide. By this time the coverslip should have touched the solution on the slide.
f] Slowly remove the forceps so that the coverslip is gently lowered into its final resting position.
g] Remove excess solution by touching the side of a Kimwipe or paper towel to the narrow edges of the coverslip. Be careful not to drag out your sections with the excess solution.
h] if you have been using a stain, add water to one end of the coverslip while withdrawing the stain at the opposite end with a Kimwipe or towel. In most cases you do not need to get all of the stain out.
i] Wipe excess fluid from the bottom of the slide or it will stick on the stage and make your life more miserable than it already is.
Stains
Phloroglucinol-HCl - This stains lignified and suberized cell
walls red-orange. The stain is colorless until |
Toluidine
Blue - This is the stain that we will use most frequently, so be sure you
learn how to use it in the first few labs. It is a metachromatic (many colors) stain, and
stains lignified walls blue-green. Unlignified walls with lots of pectin stain cherry red.
However, if you over-strain (too long) with Toluidine blue, everything will be blue. Add
several sections to a drop of water on a slide. Add a drop of Toluidine blue to this.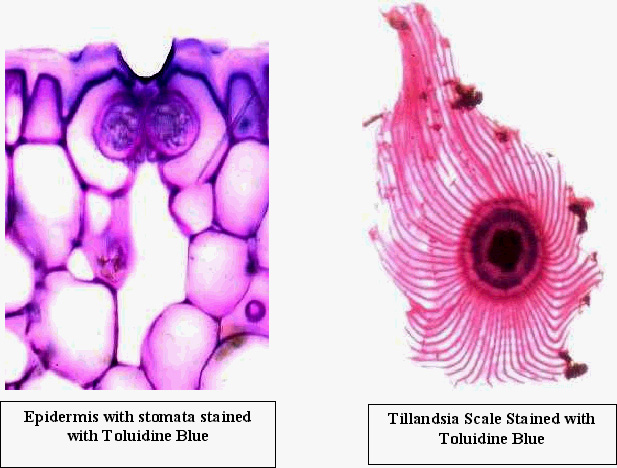
a] Quickly add a coverslip.
b] Remove the excess stain by blotting with a Kimwipe. Wipe excess fluid from the bottom of the slide.
c] View right away.
Caution
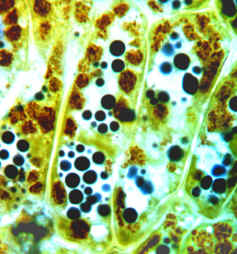
: Toluidine Blue is hard to get out of clothing, so use it carefully and clean up any spills with lots of water. In addition, it is poisonous, so avoid getting it on your skin as much as possible. We will have surgical gloves available if you want to protect your hands. Be sure to wash your hands well if they become stained.IKI - This will stain starch blue-black to orange depending on the type of starch present. It will also stain nuclei a golden color. Cell walls also stain light yellow with IKI. It is frequently not necessary to remove IKI before viewing the sections.
|
 |
Cells from Anthurium tepal stained with IKI. The brown structures are Amyloplasts.
|
Vascular bundles from Papyrus, unstained & stained with IKI |
Sudan
- This stains suberized cell walls and 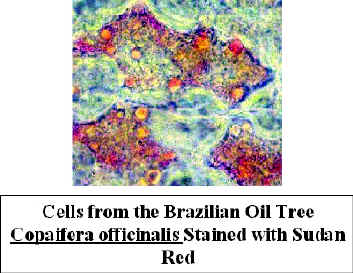 oil
in cells. The stain is dissolved in alcohol. when the specimen is rinsed with water, waxy
materials which have taken up the stain remain red, other areas are colorless. Place
several drops of stain on the slide and add sections to it. The alcohol evaporates
rapidly, so it is best to add a coverslip right away. It takes a few minutes for the stain
to work, so you will need to add stain periodically to the edge of the coverslip to
prevent the formation of air bubbles. Wash the stain out with water as described above.
Look for red-orange areas.
oil
in cells. The stain is dissolved in alcohol. when the specimen is rinsed with water, waxy
materials which have taken up the stain remain red, other areas are colorless. Place
several drops of stain on the slide and add sections to it. The alcohol evaporates
rapidly, so it is best to add a coverslip right away. It takes a few minutes for the stain
to work, so you will need to add stain periodically to the edge of the coverslip to
prevent the formation of air bubbles. Wash the stain out with water as described above.
Look for red-orange areas.
Slide Preservation - Slides can be saved for short periods by sealing the edges of the coverslip with freezing medium. This is used to stabilize tissues for cryosectioning and works well for saving slides. However, it only lasts for a day or two. Apply one generous coat and allow to dry. Then add a second coat. Staining intensity will degrade over time but slides can be saved for a long time in this state.
Polarizing Filters -
These cause light to vibrate in one plane. Light traveling along a straight line vibrates
in all possible  planes.
Imagine many radii emanating from a common center. These would represent the many
vibrational planes of the light beam. A polarizer cuts out all but one of these.
planes.
Imagine many radii emanating from a common center. These would represent the many
vibrational planes of the light beam. A polarizer cuts out all but one of these.
if two polarizers are oriented at 90o to one another, no light will pass through the second one in the series. Verify this by holding one polarizer while looking at a bright object. Take a second polarizer in your other hand and superimpose it on the first. Turn either one until the light is completely blocked.
If a crystalline or paracrystaile object is placed between crossed polarizers, it will depolarize the light which passes through it. This property is known as birefringence. Consequently, the birefringent material will be visible while all else will remain dark. Cell walls, crystals and some starch grains are birefringent, and become apparent using polarized light. This works with unstained and stained sections.