Botanik online 1996-2004. Die Seiten werden nicht mehr bearbeitet, sie bleiben als historisches Dokument der botanischen Wissenschaft online erhalten!
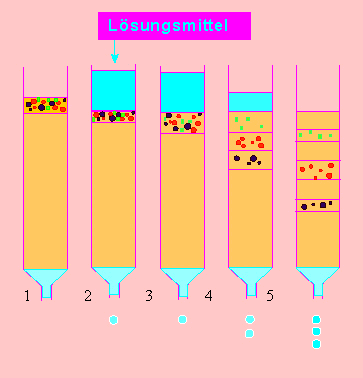
Linkes Bild: Säulenchromatographie: Auftrennung eines Molekülgemisches. bestehend (hier) aus drei Substanzen. Die Trennung beruht (wie bei der Papierchromatographie) auf unterschiedlicher Verzögerung (Retention) der Wandergeschwindigkeit durch die Packung des Säuleninhallts. Je nach Wahl des Trägermaterials wird zwischen Ionenaustauscherchromatographie, Molekülsiebchromatographie, Absorptionschromatographie u. a. unterschieden. Das durch eine Säule hindurchgewaschene Material heißt Eluat.
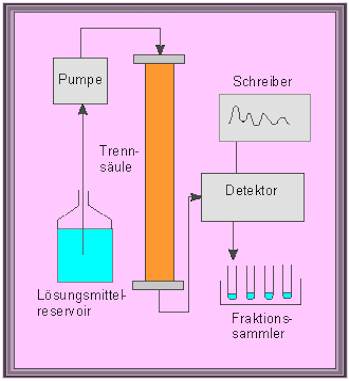
Rechtes Bild: Zusätzlich erforderliche Einrichtungen für eine automatisierte Säulenchromatographie: Schema eines Flußdiagramms.
Die Schwierigkeit, ein bestimmtes Protein in reiner Form zu gewinnen, liegt in der Tatsache begründet, daß alle Proteine aus den gleichen Bausteinen bestehen und daß Gemeinsamkeiten zwischen ihnen oft größer als die Unterschiede sind. Es gibt zumindest drei Verfahrensweisen zur Anreicherung, Isolierung und Charakterisierung spezifischer Proteine. Meist ist eine Kombination mehrerer aufeinanderfolgender Reinigungsschritte erforderlich.
![]() Ausfällung,
Ausfällung,
![]() chromatographische Auftrennung
chromatographische Auftrennung
![]() Trennung im elektrischen Feld (Elektrophorese).
Trennung im elektrischen Feld (Elektrophorese).
Der erste Schritt ist stets das Aufbrechen der Zellen. Da dabei Plasma und Vakuoleninhalt vermischt werden, sind besondere Schutzmaßnahmen erforderlich, um eine Denaturierung der Proteine zu verhindern. Der pH-Wert muß durch Verwendung eines geeigneten Puffers konstant gehalten werden, und bestimmte Zusätze sind erforderlich, um freiwerdende toxische Substanzen oder unerwünschte Enzymaktivitäten zu neutralisieren.
Eine Fällung, z.B. durch eine konzentrierte Ammoniumsulfatlösung oder durch ein organisches Lösungsmittel (z.B. Aceton) ist oft der erste Reinigungs- und Konzentrierungsschritt. Das gefällte Material wird abzentrifugiert (manchmal ist eine hochtourige Zentrifuge - eine Ultrazentrifuge - hierfür erforderlich) und anschließend in einem geeigneten Lösungsmittel wieder aufgenommen. Was aber "geeignet" ist, hängt vom jeweiligen Protein und vom Ausgangsmaterial ab. Angaben hierzu sind Praktikumsanleitungen und Laborhandbüchern zu entnehmen, sie sind auch im "Material und Methoden"-Teil einschlägiger wissenschaftlicher Publikationen enthalten. Eine chromatographische Auftrennung erfolgt nach
| unterschiedlicher Teilchengröße (Molekularsiebeffekt), | |
| unterschiedlicher Ladung (unterschiedlicher Zahl ionisierter Gruppen im Molekül: -Aminogruppen, -Karboxylgruppen) | |
| oder unterschiedlicher Affinität zum absorbierenden Material. |
Eine der am weitesten verbreiteten Methoden ist die Säulenchromatographie (s. oben). Früher weit verbreitet, heute etwas in den Hintergrund geraten ist die Papierchromatographie.

Schema der Anfertigung eines zweidimensionalen Papierchromatogramms. Nach Abschluß des ersten Laufs wird das Papier (oder ein anderer Träger) getrocknet und um 90° gedreht. Die zweidimensionale Chromatographie nutzt das unterschiedliche Lösungsvermögen der zu trennenden Substanzen in zwei verschiedenen Fließmitteln (meist Lösungsmittelgemischen) aus.
Neben der Chromatographie spielt in allen analytisch arbeitenden
Laboratorien die Elektrophorese eine wichtige Rolle. Auch hier
gibt es zahlreiche Varianten in der Ausführung. Am gebräuchlichsten
sind Trennungen nach Ladung oder nach Molekülgröße.
Letzteres ist jedoch insofern problematisch, weil das Protein
hierbei denaturiert werden muß und Enzymaktivitäten
anschließend nicht mehr meßbar sind. (Im Vorgriff
: Bei der Nukleotidsequenzierung von DNS spielt gerade diese Methode
eine herausragende Rolle. Es ist z.B. möglich, ein Molekül
aus 99 Nukleotiden von einem aus 98 oder 100 aufgrund ihrer Wandergeschwindigkeit
im elektrischen Feld zu unterscheiden.) In der Regel erfolgt die
Trennung des Trennguts (z.B. eines Proteingemisches) in einem
Trägergel (hierfür ist Polyacrylamid sehr gut geeignet);
man kann aber auch mit Papier arbeiten, oder mit einem Agarose oder Stärkegel.


Prinzip der Gelelektrophorese. Einfluß von Ladung und Teilchengröße auf die elektrophoretische Beweglichkeit von Proteinen oder anderen Makromolekülen, z. B. Nukleinsäuren. A: Trennung aufgrund von Ladungen, B: Trennung aufgrund der Teilchengröße, C. Addition von A und B, D. Kompensation von A und B.
Eine Modifikation der Gelelektrophorese ist die Isoelektrische Fokussierung. Hierbei wird außer dem elektrischen Feld ein pH-Gradient angelegt, der dafür sorgt, daß das wandernde Protein in der Nähe seines isoelektrischen Punkts (pK) zum Stehen kommt. Der isoelektrische Punkt wiederum ist durch jenen pH-Wert gekennzeichnet, an dem die geringste Zahl ionisierter Gruppen im Molekül vorhanden ist. Im sauren Bereich liegen die Amino- und die Karboxylgruppen in Form von -NH3+, bzw. -COOH vor, im alkalischen als -NH2 und -COO-, und am isoelektrischen Punkt finden wir -NH2- und -COOH-Gruppen. Es hängt daher vor allem vom Verhältnis der basischen zu den sauren Aminosäuren im Protein ab, wo sein isoelektrischer Punkt liegt; die Löslichkeit des Proteins ist hier drastisch reduziert.
Um die Reinheit eines Proteins zu definieren, können verschiedene Kriterien herangezogen werden:
Stets ist es erforderlich, mehrere voneinander unabhängige Reinheitskriterien anzugeben, denn keines ist für sich alleine ausreichend, um eine unangreifbare Aussage zu machen. Manche Proteine bilden Kristalle nur im Komplex mit andersartigen Substanzen; andere tragen auf einer Polypeptidkette zwei enzymatische Aktivitäten, oder die Aktivität ist an einen Komplex (eine Quartärstruktur) aus mehreren, z.T. verschiedenen Polypeptidketten gebunden. Eine weitere Gruppe schließlich ist, wie wir schon gesehen haben, auf Kofaktoren angewiesen. Viele der Membranproteine sind nur im Verbund mit Lipiden aktiv usw. Reinheit einer Polypeptidkette und Enzymaktivität sind folglich zwei voneinander verschiedene Größen. Man muß sich daher oft schon vor Beginn eines Experiments entscheiden, was man durch eine Reinigung erreichen will und was man mit dem Produkt anstellen möchte. Parallelansätze und das Arbeiten nach verschiedenen Strategien, gehört zum Laboralltag.
Die Ansprüche sind stetig gestiegen. Heute genügt es oft nicht mehr, ein Protein aus einer Zelle zu isolieren. Vielmehr ist man daran interessiert, dieses aus einem bestimmten Kompartiment (Zellkern, Plastiden, Mitochondrien usw.) zu gewinnen bzw. es als Bestandteil eines bestimmten Kompartiments zu identifizieren. Der Reinigung der Proteine ist damit eine Anreicherung des Kompartiments vorgeschaltet. Ein wichtiges Hilfsmittel ist dabei die Dichtegradientenzentrifugation in der Ultrazentrifuge.
Die Empfindlichkeit der Nachweismethoden kann durch gezielten Einsatz radioaktiv markierter Ausgangsverbindungen um Größenordnungen gesteigert werden.
